
Aucun produit
COMMENT FONCTIONNENT LES ESSAIS CLINIQUES ET QUI PEUT PARTICIPER?
Les essais cliniques sont des études de recherche qui visent à déterminer si une stratégie, un traitement ou un dispositif médical est sûr pour une utilisation ou une consommation humaine.
Ces études peuvent également évaluer l'efficacité d'une approche médicale pour des conditions ou des groupes de personnes spécifiques.
Dans l'ensemble, ils enrichissent les connaissances médicales et fournissent des données fiables pour aider à la prise de décisions et aux directives en matière de soins de santé.
Pour assurer la sécurité des participants, les essais commencent par de petits groupes et examinent si une nouvelle méthode cause des dommages ou des effets secondaires insatisfaisants. En effet, une technique réussie en laboratoire ou chez l'animal peut ne pas être sûre ou efficace pour l'homme.
Quelques faits sur les essais cliniques
- Les essais cliniques visent à déterminer si une stratégie, un traitement ou un dispositif médical est sûr et efficace pour les humains à utiliser ou à consommer.
- Les essais se composent de quatre phases et peuvent se concentrer sur: le traitement, la prévention, le diagnostic, le dépistage, les soins de soutien, la recherche sur les services de santé et les sciences fondamentales.
- Une équipe de recherche comprendra probablement des médecins, des infirmières, des travailleurs sociaux, des professionnels de la santé, des scientifiques, des gestionnaires de données et des coordonnateurs d'essais cliniques.
- La participation peut comporter à la fois des risques et des avantages. Les participants doivent lire et signer le & # 8220; consentement éclairé & # 8221; document avant de rejoindre un procès.
- Les risques sont contrôlés et surveillés, mais la nature des études de recherche médicale signifie que certains risques sont inévitables.
Quels sont les essais cliniques?
Le but principal des essais cliniques est la recherche. Les essais sont conçus pour enrichir les connaissances médicales liées au traitement, au diagnostic et à la prévention des maladies ou affections.

Les essais cliniques sont des études de recherche qui visent à déterminer si une stratégie, un traitement ou un dispositif médical est sûr pour une utilisation ou une consommation humaine.
Les études suivent des normes et des directives scientifiques strictes qui visent à:
- protéger les participants
- fournir des résultats fiables et précis
Les essais cliniques sur l'homme ont lieu aux dernières étapes d'un processus de recherche long, systématique et approfondi.
Le processus commence souvent dans un laboratoire, où de nouveaux concepts sont développés et testés.
Les tests sur les animaux permettent aux scientifiques de voir comment l'approche affecte un corps vivant.
Enfin, les tests sur l'homme sont effectués en petits et puis en plus grands groupes.
Des essais peuvent être effectués pour:
- Évaluer une ou plusieurs interventions de traitement pour une maladie, un syndrome ou une condition, tels que des médicaments, des dispositifs médicaux ou des approches de chirurgie ou de thérapies
- Évaluer les moyens de prévenir une maladie ou un état , par exemple, grâce à des médicaments, des vaccins et des changements de style de vie
- Évaluer une ou plusieurs interventions de diagnostic qui pourraient identifier ou diagnostiquer une maladie ou une condition particulière
- Examiner les méthodes d'identification pour reconnaître une condition ou des facteurs de risque pour cette condition
- Explorer les procédures de soins de soutien pour améliorer le confort et la qualité de vie des personnes atteintes d'une maladie chronique
Le résultat d'un essai clinique peut déterminer si une nouvelle stratégie, un nouveau traitement ou un nouveau dispositif médical:
- a un effet positif sur le pronostic du patient
- cause un préjudice imprévu
- n'a aucun avantage positif ou a des effets négatifs
Les essais cliniques peuvent fournir des informations précieuses concernant la rentabilité d'un traitement, la valeur clinique d'un test de diagnostic et la façon dont un traitement améliore la qualité de vie.
Types d'essais cliniques
Tous les essais cliniques ont un objectif principal. Celles-ci peuvent être réparties dans les catégories suivantes:
- Traitement: tester de nouveaux traitements, de nouvelles combinaisons de médicaments ou de nouvelles approches chirurgicales ou thérapeutiques
- Prévention: examiner les moyens d'améliorer la prévention ou la récurrence des maladies, par exemple, par le biais de médicaments, de vitamines , de vaccins, de minéraux et de changements de style de vie
- Diagnostic: trouver des techniques et des procédures de test améliorées pour diagnostiquer les maladies et les affections
- Dépistage: tester la meilleure méthode d'identification de certaines maladies ou conditions de santé
- Soins de soutien: Enquête sur les procédures pour améliorer le confort et la qualité de vie des patients atteints d'une maladie chronique
- Recherche sur les services de santé: évaluation de la prestation, du processus, de la gestion, de l'organisation ou du financement des soins de santé
- Science fondamentale: Examiner le fonctionnement d'une intervention
Pourquoi les essais cliniques sont-ils importants?
Les essais cliniques contribuent à améliorer et à faire progresser les soins médicaux. Les études fournissent des preuves factuelles qui peuvent être utilisées pour améliorer les soins aux patients.
La recherche clinique n'est menée que si les médecins ne connaissent pas des éléments tels que:
- si une nouvelle approche fonctionne efficacement chez l'homme et est sûre
- quels traitements ou stratégies fonctionnent le mieux pour certaines maladies et certains groupes de personnes
Comment fonctionnent les essais cliniques?
Différents éléments interviennent dans la mise en place, la conduite et le suivi d'un essai clinique.
Protocole d'essais cliniques

Un protocole est la description écrite d'un essai clinique. Il comprend les objectifs, la conception, les méthodes, le contexte scientifique et les informations statistiques de l'étude.
Un essai suit un plan ou un protocole complet. Un protocole est la description écrite d'un essai clinique.
Il comprend les objectifs, la conception et les méthodes de l'étude, le contexte scientifique pertinent et les informations statistiques.
Les informations clés à inclure peuvent être:
- le nombre de participants
- qui est éligible pour participer
- quels tests seront donnés et à quelle fréquence
- types de données à collecter
- la durée de l'étude
- des informations détaillées sur le plan de traitement
Éviter les biais
Les chercheurs doivent prendre des mesures pour éviter les biais.
Le biais fait référence aux choix humains ou à d'autres facteurs qui ne sont pas liés au protocole mais qui peuvent affecter les résultats de l'essai.
Les étapes qui peuvent aider à éviter les biais sont les groupes de comparaison, la randomisation et le masquage.
Groupes de comparaison
La plupart des essais cliniques utilisent des groupes de comparaison pour comparer les stratégies et les traitements médicaux. Les résultats montreront si un groupe a un meilleur résultat que l'autre.
Cela se fait généralement de deux manières:
- Un groupe reçoit un traitement existant pour une condition, et le deuxième groupe reçoit un nouveau traitement. Les chercheurs comparent ensuite le groupe qui a les meilleurs résultats.
- Un groupe reçoit un nouveau traitement et le deuxième groupe reçoit un placebo , un produit inactif qui ressemble au produit testé.
Randomisation
Les essais cliniques avec des groupes de comparaison utilisent souvent la randomisation. Les participants sont répartis en groupes de comparaison par hasard plutôt que par choix. Cela signifie que toute différence observée au cours d'un essai sera due à la stratégie utilisée et non à des différences préexistantes entre les participants.
Masquage ou aveuglement
Le masquage ou l'aveuglement aide à éviter les biais en n'informant ni les participants ni les chercheurs du traitement qu'ils recevront.
Seul aveugle : c'est lorsque les participants ou les chercheurs ne savent pas quel groupe est lequel.
En double aveugle : c'est lorsque les participants et les chercheurs ne sont pas au courant.
Facteurs de confusion
Un confondant peut fausser la véritable relation entre deux ou plusieurs caractéristiques.
Par exemple, on pourrait conclure que les personnes qui portent un briquet sont plus susceptibles de développer un cancer du poumon parce que le port d'un briquet provoque un cancer du poumon. Le tabagisme est un facteur de confusion dans cet exemple.
Les personnes qui portent un briquet sont plus susceptibles d'être des fumeurs et les fumeurs sont plus susceptibles de développer un cancer du poumon, mais certaines personnes peuvent porter un briquet à d'autres fins.
Ne pas en tenir compte peut conduire à de fausses conclusions.
Qui fait partie de l'équipe de recherche?
Un chercheur principal, qui est généralement un médecin, dirigera chaque étude clinique.
L'équipe de recherche peut comprendre :
- médecins
- infirmières
- les travailleurs sociaux
- professionnels de la santé
- scientifiques
- gestionnaires de données
- coordinateurs d'essais cliniques
Où sont menés les essais cliniques?
L'emplacement dépendra du type d'étude et de l'organisateur.
Certains emplacements courants incluent:
- les hôpitaux
- universités
- centres médicaux
- cabinets de médecins
- cliniques communautaires
- sites de recherche financés par le gouvernement fédéral et par l'industrie
Combien de temps durent les essais?
Cela dépend de ce qui est étudié, entre autres facteurs. Certains essais ont eu lieu les derniers jours, tandis que d'autres se poursuivent pendant des années.
Avant de s'inscrire à un essai, les participants seront informés de la durée prévue.
Conception et organisation
Il existe différents types d'études et différentes façons de les organiser. Voici quelques types d'études.
Études d'observation
Les études de cohorte et les études cas-témoins sont des exemples d'études d'observation.
Étude de cohorte

Une étude de cohorte est une étude observationnelle dans laquelle les participants sont sélectionnés et suivis dans le temps, pour voir la probabilité de développement d'une maladie au sein du groupe.
Une étude de cohorte est une étude observationnelle dans laquelle la population à l'étude, ou cohorte, est sélectionnée.
Des informations sont recueillies pour déterminer quels sujets ont soit:
- une caractéristique particulière, comme un groupe sanguin qui serait lié au développement de la maladie en question
- exposition à un facteur pouvant être lié à une maladie, par exemple le tabagisme
Un individu pourrait être choisi parce qu'il fume. Ils peuvent ensuite être suivis à temps pour voir dans quelle mesure ils sont susceptibles de développer une maladie, par rapport à d'autres personnes.
Ce type d'étude est utilisé pour étudier l'effet de facteurs de risque suspectés qui ne peuvent pas être contrôlés expérimentalement, tels que l'impact du tabagisme sur le cancer du poumon.
Les principaux avantages des études de cohorte sont:
- L'exposition est mesurée avant le début de la maladie et est donc susceptible d'être impartiale en termes de développement de la maladie.
- Les expositions rares peuvent être étudiées par une sélection appropriée de cohortes d'étude.
- Plusieurs résultats - ou maladies - peuvent être étudiés pour une même exposition.
- L'incidence de la maladie peut être calculée à la fois dans les groupes exposés et non exposés.
Les principaux inconvénients des études de cohorte sont:
- Ils ont tendance à être coûteux et longs, surtout s'ils sont menés de manière prospective, ce qui signifie aller de l'avant.
- Les changements dans l'état d'exposition et les critères de diagnostic au fil du temps peuvent affecter la classification des individus en fonction de l'exposition et de l'état de la maladie.
- Il pourrait y avoir un biais d'information dans le résultat final car le statut d'exposition du sujet est connu.
- Les pertes de suivi peuvent présenter un biais de sélection.
Études cas-témoins
Une étude cas-témoins peut distinguer les facteurs de risque d'une condition médicale particulière.
Les chercheurs comparent les gens avec une condition et ceux sans elle. En remontant dans le temps, ils identifient la différence entre les deux groupes.
Les études cas-témoins sont toujours rétrospectives - rétrospectives - car elles commencent par le résultat puis remontent pour enquêter sur les expositions.
Les principaux avantages des études cas-témoins sont:
- Les résultats peuvent être obtenus rapidement.
- L'étude peut avoir lieu avec un minimum de financement ou de parrainage.
- Ils sont efficaces pour étudier les maladies rares ou les maladies avec une longue période d'induction.
- Un large éventail de facteurs de risque possibles peut être examiné.
- Plusieurs expositions peuvent être étudiées.
- Ils nécessitent peu de sujets d'étude.
Les principaux inconvénients des études cas-témoins sont:
- Les données d'incidence ne peuvent pas être générées.
- Ils sont sujets à des biais.
- Il peut être difficile d'obtenir des mesures précises et impartiales des expositions passées si la tenue des registres est inadéquate ou peu fiable. C'est ce qu'on appelle le biais d'information.
- La sélection des contrôles peut être problématique. Cela peut introduire un biais de sélection.
- La séquence chronologique entre l'exposition et la maladie peut être difficile à identifier.
- Ils ne conviennent pas pour l'examen d'expositions rares, sauf si l'exposition est responsable d'un pourcentage élevé de cas.
Étude cas-témoins imbriquée
Dans une étude cas-témoins imbriquée, les groupes - cas et témoins - proviennent de la même population ou cohorte d'étude.
À mesure que la cohorte est suivie, les cas qui surviennent deviennent les «cas» dans l'étude cas-témoins. Les participants non affectés de la cohorte deviennent les «témoins».
Les études cas-témoins imbriquées sont moins coûteuses et prennent moins de temps par rapport à une étude de cohorte.
Les taux d'incidence et de prévalence de la maladie peuvent parfois être projetés à partir d'une étude de cohorte cas-témoins imbriquée. Cela n'est pas possible à partir d'une simple étude cas-témoins, car le nombre total d'individus exposés et les délais de suivi sont généralement inconnus.
Les principaux avantages des études cas-témoins imbriquées sont les suivants:
- Efficacité: tous les participants de la cohorte ne nécessitent pas de tests diagnostiques.
- Flexibilité: Ils permettent de tester des hypothèses qui n'étaient pas anticipées lors de la planification de la cohorte.
- Réduction du biais de sélection: les cas et les témoins sont échantillonnés dans la même population.
- Réduction du biais d'information: l'exposition aux facteurs de risque peut être évaluée avec l'investigateur aveugle au statut du cas.
Le principal inconvénient est que les résultats ont une autorité inférieure, en raison de la petite taille de l'échantillon.
Etude écologique
Une étude écologique examine la relation entre l'exposition et les résultats de la population ou de la communauté.
Les catégories communes d'études écologiques comprennent:
- comparaisons géographiques
- analyse des tendances temporelles
- études de migration
Les principaux avantages des études écologiques sont:
- Ils sont peu coûteux, car les données de santé collectées régulièrement peuvent être utilisées.
- Ils prennent moins de temps que d'autres études.
- Ils sont simples et faciles à comprendre.
- L'effet des expositions mesurées sur des groupes ou des zones - comme le régime alimentaire, la pollution de l'air et la température - peut être étudié.
Les principaux inconvénients des études écologiques sont:
- Des erreurs de déduction appelées erreur écologique peuvent se produire. Cela se produit lorsque les chercheurs tirent des conclusions sur les individus uniquement sur la base de l'analyse des données de groupe.
- L'exposition aux relations de résultats est difficile à détecter.
- Il y a un manque d'informations sur les facteurs de confusion.
- Il peut y avoir des différences systématiques entre les domaines dans la façon dont les expositions sont mesurées.
Etudes expérimentales
Outre les études observationnelles, il existe également des études expérimentales, y compris des études de traitement.
Essais contrôlés randomisés

Un essai contrôlé randomisé (ECR) répartit au hasard les individus pour qu'ils reçoivent ou non une intervention particulière.
Un des deux traitements différents sera utilisé, ou un traitement et un placebo.
Il s'agit du type d'étude le plus efficace pour identifier le traitement qui fonctionne le mieux. Il réduit l'influence des variables externes.
Les principaux avantages des ECR sont:
- Il n'y a pas de parti pris conscient ou subconscient de la part du chercheur. Cela garantit essentiellement la validité externe.
- Des variables de confusion telles que l'âge, le sexe, le poids, le niveau d'activité, etc., peuvent être annulées tant que le groupe d'échantillonnage est suffisamment grand.
Les principaux inconvénients des ECR sont:
- Ils prennent du temps.
- Ils peuvent être chers.
- Ils nécessitent de grands groupes d'échantillons.
- Les événements rares peuvent être difficiles à étudier.
- Des erreurs statistiques faussement positives et faussement négatives sont possibles.
Essai clinique adaptatif
Une méthode de conception adaptative est basée sur les données collectées. Il est à la fois flexible et efficace. Des modifications peuvent être apportées à l'essai et aux procédures statistiques des essais cliniques en cours.
Quasi-expérience
Les études quasi-expérimentales ou «non randomisées» comprennent un large éventail d'études d'intervention qui ne sont pas randomisées. Ce type d' essai est fréquemment utilisé lorsqu'un ECR n'est pas réalisable ou éthique sur le plan logistique.
Hiérarchie des preuves
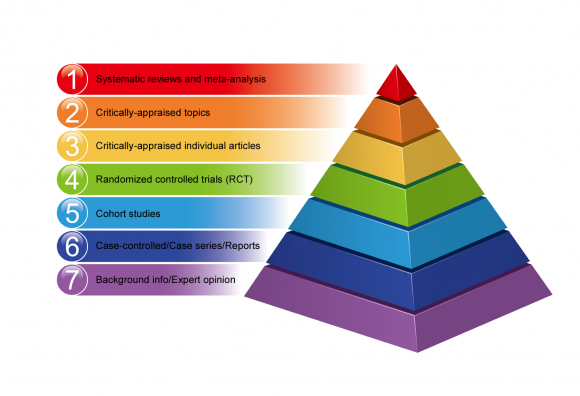
Un certain nombre de hiérarchies de preuves ont été établies pour permettre de classer diverses méthodes de recherche selon la validité de leurs résultats.
Des hiérarchies de preuves permettent de classer différentes méthodes de recherche en fonction de la validité de leurs résultats.
Tous les plans de recherche ne sont pas égaux en termes de risque d'erreur et de biais dans leurs résultats. Certaines méthodes de recherche fournissent de meilleures preuves que d'autres.
Vous trouverez ci-dessous un exemple de la hiérarchie de la médecine factuelle sous la forme d'une pyramide , allant d'une preuve de qualité inférieure en bas à une preuve de haute qualité en haut.
Phases d'un essai clinique
Les études de recherche médicale sont divisées en différentes étapes, appelées phases. Pour les tests de dépistage de drogues, ceux-ci sont définis par la FDA.
Les premiers essais cliniques étudient l'innocuité d'un médicament et les effets secondaires qu'il peut provoquer. Des essais ultérieurs testent si un nouveau traitement est meilleur qu'un traitement existant.
Essais de phase 0: pharmacodynamique et pharmacocinétique
La phase 0 est une phase exploratoire qui permet de fournir des informations cliniques sur un nouveau médicament à une phase antérieure.
- est réalisée au début de la phase 1
- implique une exposition humaine très limitée
- n'a aucune intention thérapeutique ou diagnostique, se limitant aux études de dépistage et de microdose
Essais de phase 1: dépistage de la sécurité
Après la phase 0, il y a quatre autres phases d'essais chez l'homme. Celles-ci se chevauchent souvent. Les phases 1 à 3 ont lieu avant l'octroi d'une licence.
Les directives de la phase 1 impliquent:
- entre 20 et 80 volontaires sains
- vérification des effets secondaires les plus fréquents du médicament
- découvrir comment le médicament est métabolisé et excrété
Essais de phase 2: établir l'efficacité
Si les études de phase 1 ne révèlent pas de niveaux de toxicité inacceptables, les études de phase 2 peuvent commencer.
Cela implique:
- entre 36 et 300 participants
- recueillir des données préliminaires sur l'efficacité du médicament chez les personnes atteintes d'une certaine maladie ou affection
- essais contrôlés pour comparer ceux qui reçoivent le médicament avec des personnes dans une situation similaire qui reçoivent un médicament différent ou un placebo
- évaluation continue de la sécurité
- études des effets secondaires à court terme
Essais de phase 3: confirmation finale de l'innocuité et de l'efficacité
Si la phase 2 a confirmé l'efficacité d'un médicament, la FDA et les sponsors discuteront de la manière de mener des études à grande échelle au cours de la phase 3.
Cela impliquera:
- entre 300 et 3000 participants
- collecte d'informations supplémentaires sur la sécurité et l'efficacité
- études de différentes populations
- examiner divers dosages pour déterminer le meilleur montant d'ordonnance
- utiliser le médicament en combinaison avec d'autres médicaments pour déterminer l'efficacité
Après cette phase, les informations complètes sur le nouveau médicament sont transmises aux autorités sanitaires.
Réunion d'examen
Si la FDA approuve la commercialisation du produit, des études sur les exigences et l'engagement post-commercialisation sont effectuées.
La FDA utilise ces études pour collecter des informations supplémentaires sur l'innocuité, l'efficacité ou l'utilisation optimale du produit.
Demande de nouveau médicament

Un promoteur de médicament remplira une demande de nouveau médicament (NDA) pour demander à la FDA d'envisager d'approuver un nouveau médicament pour la commercialisation aux États-Unis.
Un NDA comprend:
- toutes les données animales et humaines
- analyse de données
- informations sur le comportement des drogues dans le corps
- détails de fabrication
La FDA dispose de 60 jours pour décider de la déposer ou non pour examen.
S'ils décident de déposer la NDA, l'équipe d'examen de la FDA est chargée d'évaluer les recherches du promoteur sur l'innocuité et l'efficacité du médicament.
Les étapes suivantes doivent alors avoir lieu.
Étiquetage des médicaments : la FDA examine l'étiquetage professionnel du médicament et confirme que les informations appropriées sont partagées avec les consommateurs et les professionnels de la santé.
Inspection des installations : La FDA inspecte les installations où le médicament sera fabriqué.
Approbation des médicaments: les examinateurs de la FDA approuvent la demande ou émettent une lettre de réponse.
Essais de phase 4: études en cours de vente
Les essais de phase 4 ont lieu après l'approbation de la commercialisation du médicament. Ils sont conçus pour inclure:
- plus de 1000 patients
- expérience approfondie de l'évaluation de l'innocuité et de l'efficacité du nouveau médicament dans un groupe plus large et des sous-populations de patients
- comparaison et combinaison avec d'autres traitements disponibles
- évaluation des effets secondaires à long terme du médicament
- détection d'événements indésirables moins fréquents
- rapport coût-efficacité de la pharmacothérapie par rapport à d'autres thérapies traditionnelles et nouvelles
Rapport de sécurité
Une fois que la FDA a approuvé un médicament, la phase de post-commercialisation commence. Le sponsor, généralement le fabricant, soumet des mises à jour de sécurité périodiques à la FDA.
Qui parraine les essais cliniques?
Les essais cliniques et la recherche peuvent coûter des centaines de millions de dollars. Les groupes qui financent les essais peuvent inclure:
- sociétés pharmaceutiques, biotechnologiques et d'appareils médicaux
- centres médicaux universitaires
- groupes et fondations bénévoles
- Instituts nationaux de la santé
- départements du gouvernement
- médecins et prestataires de santé
- personnes
Qui peut participer?
Le protocole définit qui peut participer à un essai.
Les critères d'inclusion possibles peuvent être:
- avoir une maladie ou une condition spécifique
- être «en bonne santé», sans problème de santé
Les critères d'exclusion sont les facteurs qui empêchent certaines personnes de participer à un essai.
Les exemples incluent l'âge, le sexe, un type ou un stade spécifique d'une maladie, les antécédents de traitement et d'autres conditions médicales.
Avantages et risques possibles
La participation à des essais cliniques peut présenter à la fois des avantages et des risques pour les participants.
Les avantages possibles des essais cliniques sont les suivants:
- Les participants ont accès à de nouveaux traitements.
- Si un traitement s'avère efficace, les participants seront parmi les premiers à en bénéficier.
- Les participants qui ne font pas partie du groupe recevant un nouveau traitement peuvent recevoir le traitement standard pour l'affection particulière, qui peut être aussi bon ou meilleur que la nouvelle approche.
- La santé est étroitement surveillée et soutenue par une équipe de prestataires de santé.
- Les informations recueillies lors des essais cliniques enrichissent les connaissances scientifiques, peuvent aider les autres et, en fin de compte, améliorer les soins de santé.
Les risques possibles incluent:
- Les soins standard pour une condition particulière peuvent parfois être meilleurs que la nouvelle stratégie ou les nouveaux traitements à l'étude.
- La nouvelle approche ou le nouveau traitement peut bien fonctionner pour certains participants, mais pas nécessairement pour d'autres.
- Il peut y avoir des effets secondaires inattendus ou imprévus, en particulier dans les essais de phase 1 et de phase 2 et avec des approches telles que la thérapie génique ou de nouveaux traitements biologiques.
- L'assurance maladie et les prestataires de soins de santé ne couvrent pas toujours les soins aux patients et les coûts pour ceux qui participent aux essais cliniques.
Que signifie donner son consentement?

Les participants doivent lire attentivement le document de consentement, décider s'ils souhaitent s'inscrire et signer avant de pouvoir participer à l'essai.
Le document de consentement éclairé explique les risques et les avantages potentiels de la participation à un essai clinique.
Les éléments qui doivent apparaître dans le document comprennent, entre autres:
- but de la recherche
- risques prévisibles d'inconfort
- avantages possibles
Les participants doivent lire attentivement le document de consentement , décider s'ils souhaitent s'inscrire et signer avant de pouvoir participer à l'essai.
Les essais cliniques sont-ils sûrs?
La FDA veille à ce que toute personne qui envisage de participer à un essai ait accès à toutes les informations fiables dont elle a besoin pour faire un choix éclairé, y compris des informations sur les risques.
Bien que les risques pour les participants soient contrôlés et surveillés, certains risques peuvent être inévitables, en raison de la nature des études de recherche médicale.
Comment les participants sont-ils protégés?

Les bonnes pratiques cliniques (BPC) sont définies comme une norme pour la conception, la conduite, la performance, la surveillance, l'audit, l'enregistrement, l'analyse et le rapport des essais ou études cliniques.
La sécurité des participants est une question hautement prioritaire. Dans chaque essai, la surveillance scientifique et les droits des patients contribuent à leur protection.
Les bonnes pratiques cliniques (BPC) visent à garantir le respect des procédures éthiques et appropriées lors des essais.
La conformité aux BPC donne au public l'assurance que la sécurité et les droits des participants sont protégés.
Il vise à:
- protéger les droits, la sécurité et le bien-être des participants
- garantir la fiabilité, l'intégrité et la qualité des données collectées
- fournir des lignes directrices et des normes pour la conduite de la recherche clinique
Les fondements du GCP ont été posés pour la première fois en 1947. Les principaux points étaient que, lors de tout essai, les chercheurs doivent garantir:
- participation volontaire
- consentement éclairé
- minimisation des risques
Au fil du temps, les ajouts ont varié, allant de l'établissement d'une protection supplémentaire pour les populations vulnérables à la fourniture d'orientations aux organismes menant des recherches.
Droits des patients
Les moyens de protéger les droits des patients sont les suivants:
Le consentement éclairé est le processus consistant à fournir aux participants à l'essai clinique tous les faits concernant l'essai. Cela se produit avant que les participants n'acceptent de participer et au cours de l'essai. Le consentement éclairé comprend des détails sur les traitements et les tests qui peuvent être reçus et les avantages et risques possibles.
Autres droits : Le document de consentement éclairé n'est pas un contrat; les participants peuvent se retirer de l'étude à tout moment, que l'essai soit terminé ou non.
Droits et protection des enfants: Un parent ou un tuteur légal doit donner son consentement légal si l'enfant est âgé de 18 ans ou moins. Si un essai peut comporter un risque supérieur au minimum, les deux parents doivent donner leur autorisation. Les enfants de plus de 7 ans doivent accepter de participer aux essais cliniques.
Comment trouver un essai clinique?
Un test CRP peut être utilisé pour diagnostiquer les maladies auto-immunes inflammatoires. Faites-vous vérifier à la maison avec LetsGetChecked et recevez les résultats en ligne en 2 à 5 jours. Commandez aujourd'hui avec la livraison gratuite et 20% de réduction.